Cleaver Manual Version 04
1. Introduction
2. Installation and configuration
3. Interface
4. Data and files
5. Limitations
6. Examples
7. Functions by menu items
8. Troubleshooting
9. References
1. Introduction
Cleaver is a software tool for for finding differences in restriction endonuclease digests of orthologous DNA fragments. It is intended as a tool for molecular biologists to find endonuclease recognition sites that are specific to given alleles, populations, species or higher taxa. The main reason to use it is this ability to analyse many sequences for restriction digestion patterns at one time. As well as being capable of comparative analysis of restriction digestion, Cleaver can analyse single sequences to find restriction sites and produce restriction maps in the same way that other molecular biology software packages can.
2. Installation and configutation
Cleaver will run on Unix-like platforms such as MacOSX and Gnu/Linux systems. It is primarily developed on a GNU/Linux system, so this is likely to be the best implemented version. A version for Windows operating systems is also available. The Unix-like systems may require software on which Cleaver depends to be installed before it can run. Details of these dependencies will be given on the Cleaver home page. The Windows and MacOSX versions will be distributed as a single executable file with all the necessary software and files bundled into a convenient single folder.
Two additional programs are required for full functionality of Cleaver. One is clustalw (Thomson et al., 1994), which aligns DNA sequences for Cleaver. This is re-distributed with permission from its authors in the Windows and MacOSX packages of Cleaver, but should be installed independently for the Linux version. A web browser like Mozilla Firefox, Apple Safari, or Microsoft Internet Explorer and access to the internet is also necessary for viewing this manual. Cleaver opens a web browser to link to part of it web site that includes the manual for the program. The manual is maintained online so that it can be edited and updated independently of the program that is installed of people's computers. However, if you wish to work offline, the manual can be saved to your computer and viewed through any web browser or other software that can display html files.
3. Interface
The main interface for Cleaver is a split screen with a list of endonucleases in the top window and a window for listing sequences below that. The endonucleases are loaded automatically from a file 'EnzymeLbrary' that should be in the same directory as the Cleaver programme. Enzymes can be added or subtracted from this file by the user, or more recent versions of the whole list can be downloaded from the Cleaver site. These will be based on the files released by the Restriction Enzyme Database. Information on sequences for analysis are shown in the bottom half of the split main screen.
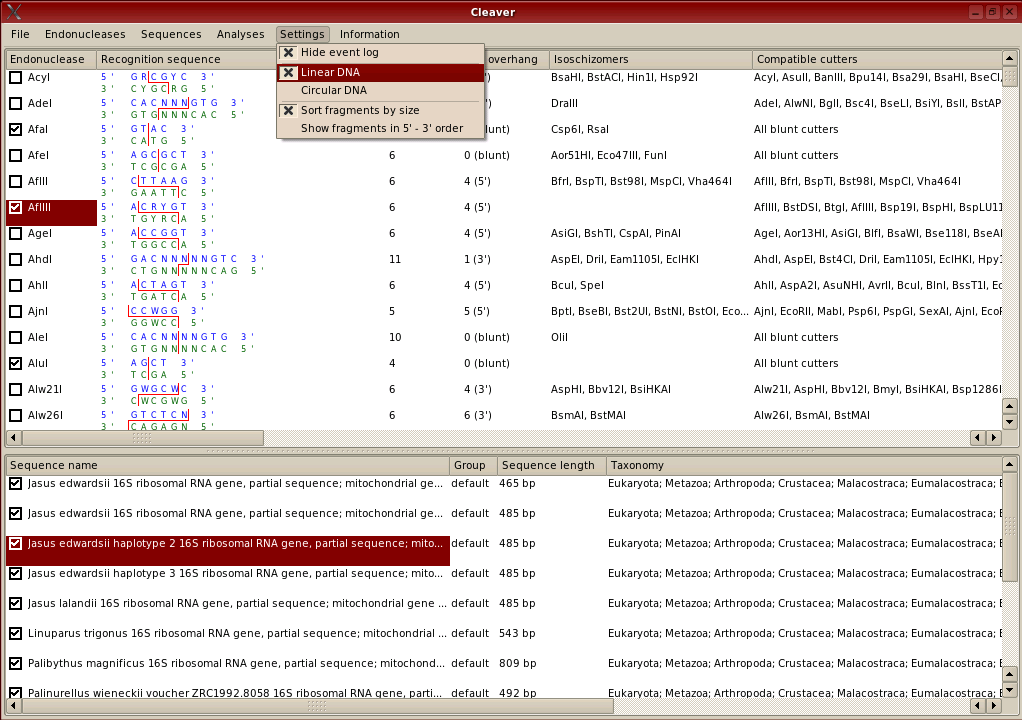
The lists of information about endonucleases and sequences are sortable. To sort the entire endonuclease window by 'Site length' for example, the user can click on the 'Site length; heading at the top of the column and the rows will be sorted in order of site length. By default, both windows are ordered by the leftmost column, which is the name of the endonuclease or sequence. The columns are also dragable, so if you want to change their order, click on the column title, hold down the left mouse button and drag the column left or right to a new position.
For any of the analyses available in Cleaver, the sequences that will be analysed and the endonucleases that will be used in the analysis will be ones that the user has selected. Selections are indicated by a tick in the box adjacent to the name of the endonuclease or sequence. To place ticks in the boxes, they can be directly indicated with the mouse pointer and left clicked or selected using options in the 'endonucleases' or 'sequences' menus. Some of these options include selecting ranges, which can be highlighted, also by using the mouse pointer.
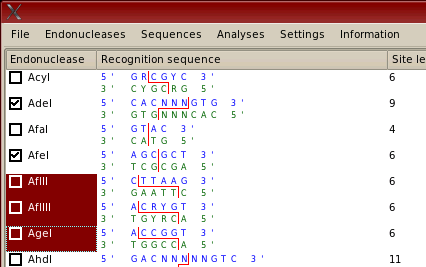
This may seem confusing, so the difference between highlighting and selection is shown below. The endonuclases 'AdeI' and 'AfeI' have been selected and will be used in any analyses. The endonucleases 'AflII', 'AflIII' and 'AgeI' have been highlighted. This means that the user can then select from the 'Endonucleases' menu options for selecting these three highlighted enzymes such as selecting (ticking) these enzymes; or for selecting others that produce 'sticky' ends compatible with cuts made by these three enzymes. Selection and highlighting of sequences is performed in the same way. Selection indicated by a tick in the checkbox indicates that an item will be used in analyses.
Analyses are selected from a pull-down menu at the top of the screen. For all analyses, the endonucleases used and the sequences analysed are only those that have been selected and have a tick in the checkbox left of their name. Sequences are all selected by default when loaded from their file. By default, no endonucleases are selected when the programme starts. The user must select some endonucleases and load some DNA sequences from a file before they can perform any analyses.
Results of analyses are given in separate windows. The text based results are copyable to the clipboard of all supported operating systems so that parts of the result output can be copied and pasted into other applications that handle text. The entire contents of results windows can also be quickly saved as a text (.txt) file, or a .png file for graphical results, using the button at the bottom left of each results window.
4. Data and Files
To use Cleaver, the user provides a set of sequences from an orthologous DNA region derived from a range of species of interest. The DNA can be stored either as FASTA, clustal, MEGA or an INSDSeq XML file.
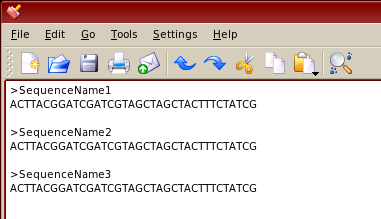
FASTA files have the simple format shown to the right. Sequence names start with a '>' character and end with a carriage return (press of the 'Enter') key. The sequence then follows until its end, which is indicated by another carriage return.
FASTA files can be downloaded from the www pages of GenBank (or other databases) or assembled in any text editor or word processor, but they have to be saved as a text file, not as a word processor file. They can be saved with the file extensions '.fas', '.fasta' or '.txt' for automatic recognition by Cleaver, or with no extension in which case they will be visible in the 'all files' option. The image to the right was assembled in a text editor for Linux and similar operating systems. For Windows systems, 'Notepad' is the best application and 'SimpleText' is the preferred option for MacOSX.
Clustal and MEGA files are produced by the programs clustalw (or clustalx) and MEGA respectively. Their format is similar to FASTA files and the ability to import data in these formats is included as a convnience in case the user has datasets in these formats already. The clustal (.aln) files allow aligned sequences to be saved for later viewing and editing in Cleaver, which may be useful in some cases.
When saving sequences taken from the databases of the NCBI (GenBank), the DNA Database of Japan, or the European Molecular Biology Organisation, INSDSeq XML files are a convenient option as they also include information on the taxonomy of the organism from which sequences are taken. This is useful in the process of making comparisons of the restriction patterns found among different taxonomic groups. To save sequences in this format, choose the 'INSDSeq XML' option from the pulldown 'display menu' as shown in the example below:

The advantage of using the INSDSeq XML format is that it includes more information associated with the sequence data than FASTA files or other common DNA file formats, which tend to only have the DNA sequence and a name associated with it,. The most important information is the taxonomic affiliation of the organism from which the DNA sequences were derived. This can be very useful for finding endonucleases specific to the given DNA region in a specific taxon.
5. Limitations
Cleaver has some limitations on display of long sequences or large numbers of graphical results. In general, sequences longer than 1200 bp may not display. They can still be manipulated and analysed, but will not show up in the sequence editor. To increase the length of sequence that is displayable on your system, you can reduce the size of the fonts and spacings used to display sequences in the 'preferences' dialog.
Cleaver can only display a limited number of graphical restriction maps at one time. The programme will not crash if you try to display too many, but the resulting window will be empty. In general, Cleaver can only display about fifteen graphical restriction maps at one time. The Windows version of the graphics library was produced by a circuitous route and is slightly buggy. Due to a bug in this library, graphical restriction maps are unavailable in the Windows version of Cleaver.
These limitations are unfortunately part of the graphical library upon which cleaver is based (PyQT version 3). A new version of this library is in development and future versions may allow larger pixmaps, which will probably allow this problem to be circumvented.
The MacOSX version of Cleaver cannot automatically link to this online documentation. For some reason, the system for passing command line arguments to another program running in a new process that works in Linux will not work on MacOSX. The MacOSX version opens a web browser, but cannot automatically direct it to this document. If any Mac programmers can help solve this problem, please email the author on simon.jarman@aad.gov.au.
6. Examples
6.1 Finding a group-specific endonuclease
Here is an example of how to identify an endonuclease that cleaves DNA from one group of organisms, but doesn't cleave the orthologous region from another group of organisms. Cleaver comes with some example files. One of them is for a set of of mitochondrial LSU rDNA sequences from the dominant group of Antarctic fishes, the Nototheniidae, 'Example1_NototheniidFish_16S.txt.' This file is in FASTA format, so it only includes name and sequence information.
The 'patagonian toothfish' Dissostichus eleginoides and its near relative Dissostichus mawsoni are members of the Nototheniidae, which are the dominant group of Antarctic fish. Imagine that you wanted to develop a simple RFLP assay for identifying Dissostichus tissue for fisheries monitoring purposes. The steps to take are as follows:
1. Open the file.
2. Manually assign a group to the two target species by selecting the appropriate option from the 'sequences' menu:
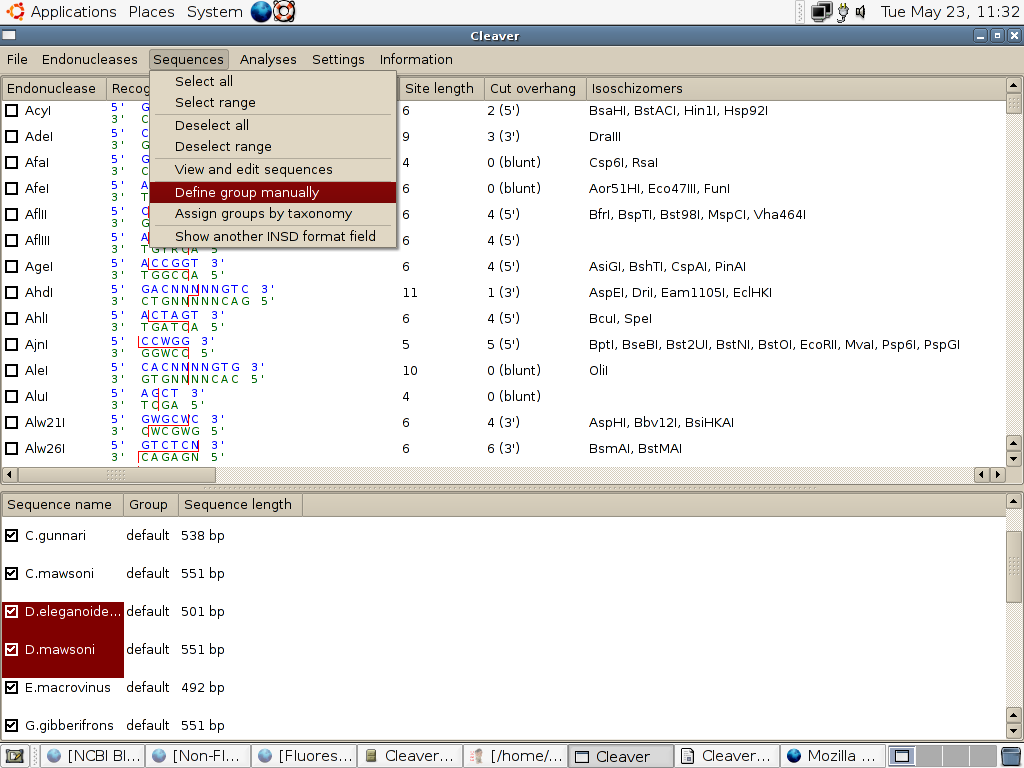
which will open a dialog for you to enter the name (which is arbitrary) for the group.
3. Select the range of endonucleases that you want to search using options in the 'Endonucleases' menu. In this case, let us select all endonucleases. Under other circumstances, you may wish to only search 4-base cutters, or select the endonuceases that you have in your laboratory.
4. From the analyses menu, select the option 'search for group-specific endonucleases.' This brings up a dialog window that allows you to define which group of sequences is to be cut and which is not to be cut. Select the groups in the left hand box and use the buttons to assign the to 'Set A' which is displayed in the top right box, or 'Set B' which is displayed in the bottom right box. The proportion of members of the sets that is acceptable to be cut / not cut may be altered. In this case, we would like an endouclease that cuts all Dissostichus DNA, but none from its near relatives.
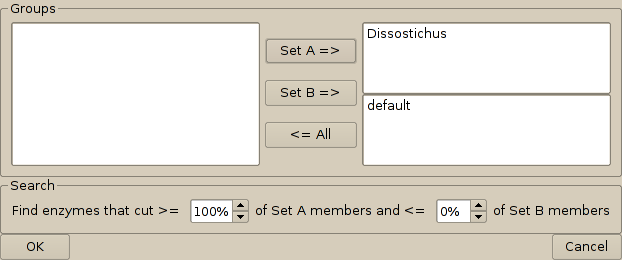
5. On pressing the 'OK' button, Cleaver searches all of the endonucleases that you selected originally to find which ones match these criteria. In this case, the result is:
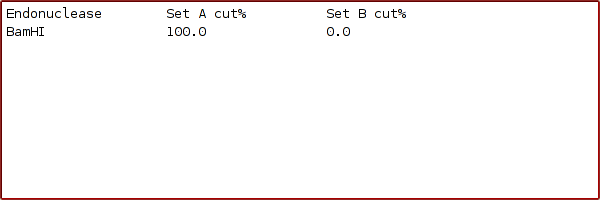
which means that you could use a BamHI digest of this DNA region to identify Dissostichus,excluding the other nototheniid fish tested as possibilities. Clearly, empirical tests to ensure that all Dissostichus have this site and all other nototheniids don't have it would be needed for confirmation.
6.2 Trimming alignments to include only a PCR generated region
Often DNA sequence information in databases is for a larger region than is convenient to analyse. It is useful to be able to remove residues from aligned DNA sequences so that the data represents the region produced by PCR or another method. In this example, imagine that you have two PCR primers:
Forward primer GGACRTGTGGCGCAYGGG
Reverse primer CCAAGCAACCCGACTC
that bind to a region of the nuclear large subunit rDNA of squids. A more extensive region of the nuclear large subunit rDNA of squid is included in the example file 'Example2_Squid_28S.gbc' which is an INSDSeq xml file downloaded directly from GenBank. This file contains not only names and DNA sequences, but a host of extra metadata relevant to the sequences.
To isolate the region of squid DNA that these primers would amplify in a PCR, the steps are:
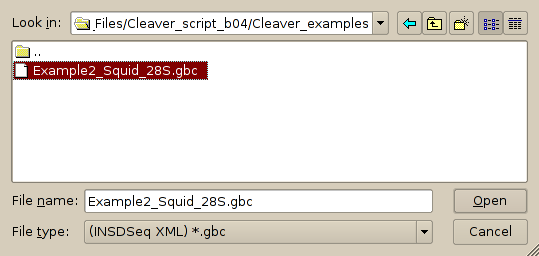
1. Open the file. This involves changing the file selector part of the file dialog to '.gbc':
2. From the 'Sequences' menu select 'View and Edit sequences', which will bring up the sequence editor:
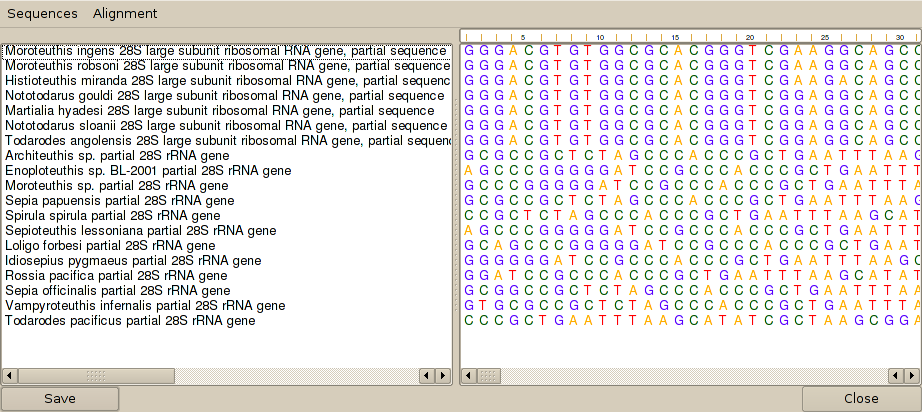
3. The sequences need to be aligned, so from the 'Alignment' menu of the sequence editor, select 'Align sequences.' This brings up a dialog that allows alignment parameters to be definded. The default parameters are generally fine, so press the 'Align' button. After a wait, Cleaver will then display the sequences in aligned format:
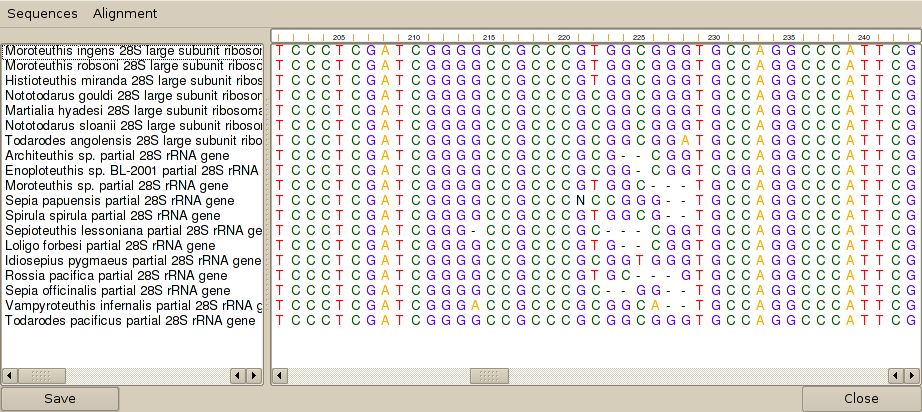
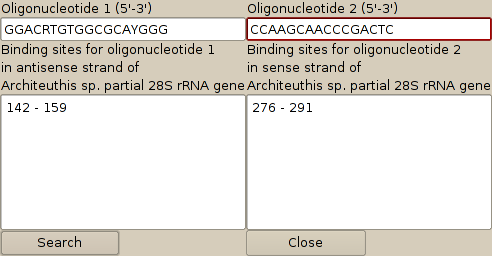
4. To find the primer binding sites, select one of the sequences (Architeuthis sp. in this case) and then select 'Search for oligonucleotide binding sites' from the 'Sequences' menu of the sequence editor. This brings up another dialog, in which the primers to search for may be entered. Pressing the 'Search' button will bring up a list of binding sites for each oligonucleotide. Note that oligonucleotides may be degenerate and that the algorithm will search across gapped positions in the alignment. The results of this search are:
5. To remove the extraneous columns in the alignment, select 'Remove columns' from the 'Alignment' menu of the sequence editor. You can then remove the columns 291-end and repeat this to remove 0-142, which will give an alignment that only includes the PCR generated region.
6. To save the changes, exit the sequence editor by pressing the 'Save' button at its bottom left hand side. The sequences are then available in their edited form for any of Cleaver's analyses. Selecting 'Close' will close the editor and discard any changes.
6.3 Displaying extra INSDSeq XML file metadata
The INSDSeq XML format contains a lot of information related to each DNA sequence in the file. By default, only five fields are displayed by Cleaver. The fileds that are displayed for the included example file 'Example2_Squid_28S.gbc' are shown below:

To display another field, select 'Display another INSDSeq format field' from the 'Sequences' menu. This will then bring up a dialog listing other fields that are available. In the example below, the creation date for the file in GenBank was selected. Cleaver has then added another column to the display showing the creation date of each sequence.

7. Functions by menu items
7.1 File
The 'File' menu provides options for loading sequence data.
7.1.1 Load sequences from file. This option opens a file opening dialogue that allows the user to select a file containing sequences for analysis. There are filters for viewing either .fas (FASTA files), .txt (Text files, assumed to contain sequences in FASTA format), or files of any type in case your file is not identified by an extension. Cleaver can only read files in four formats, FASTA, clustal, MEGA and GenBank seqXML.
7.1.2 Save sequences to file. Sequences in memory can be saved in a variety of formats, including FASTA, clustal, MEGA, PAUP and Phylip. If the sequences were originally loaded from a GenBank seqXml file, they can be saved in this format, but this format does not convert to any of the other formats or vice versa as the seqXML format contains more information associated with the sequences that is not supported by other file formats.
7.1.3 Exit. This causes Cleaver to close.
7.2 Endonucleases
The 'Endonucleases' menu lists functions for managing selection of endonucleases to be used in analyses and for managing the library of endonucleases.
7.2.1 Select all. This selects all available endonucleases for use in any analysis.
7.2.2 Select range. This selects the range of endonucleases highlighted by the user. Selection of a range can be done with the left mouse button. Use the mouse cursor to point to the first item in the range, then click the left mouse key and move the cursor while still holding down the key, which is to be released when the end of the range is reached. Alternatively, the shift key can be pressed when pointing to the first item in the range and held down until the last item in the range is left-button selected with the mouse cursor. A further alternative is to use the control ('ctrl') key, which allows similar selection to that offered by the shift key option, but the range does not need to be contiguous.
7.2.3 Select n base cutters. This will open a new dialog that allows the user to select endonucleases with cut sites that fall within a given range. the default range is 4 - 6 base sites and it can extend to the maximum length of enzyme in Cleaver's library (19 at the time of writing), while the minimum is 4.
7.2.4 Select endonucleases with cut sites compatible with highlighted endonucleases. Each endonuclease has a list of endonucleases that produce cuts that are compatible with it in the lists on the main interface. This option selects these endonucleases for analysis when the original endonuclease is highlighted.
7.2.5 Select endonucleases with names contained in 'clipboard' text. Cleaver can read the system-wide clipboard of the computer on which it is running. If the user has a text document contains endonuclease names, the names can be copied to the clipboard. This option then makes Cleaver search that text for matches to names of available endonucleases. If they are present, they will be selected for use in analyses.
7.2.6 Search library for endonucleases with a given recognition sequence. This allows the user to find endonucleases that cut a sequence. The sequence is provided and after the search button is pressed, a list of endonucleases that cut that sequence is returned. The user then highlights the endonucleases they want selected or deselected and these operations can be performed with the buttons at the bottom of the dialog.
7.2.7 Deselect all. This deselects all endonucleases (removes the tick from their checkboxes).
7.2.8 Deselect range. This deselects the endonucleases highlighted by the user.
7.2.9 Deselect isoschizomers of highlighted endonucleases. This option allows the user to search with only one endonuclease that recognises a given site. Once this endonculease (or range of endonucleases) is highlighted, any endonucleases that also recognise that site will be deselected.
7.2.10 Deselect all endonucleases with degenerate cut sites. This option deselects all available endonucleases that recognise more than one cut site and therefore have degenerate recognition sequences (e.g. AcyI – CRCGYC).
7.2.11 Add endonuclease to library. New endonucleases can be added to the library of available endonucleases with this option. It brings up a dialog window that prompts the user for an enzyme name and it's 5' – 3' recognition sequence with the point at which the sense strand is broken indicated by a '!'
7.2.12 Remove endonucleases from library. This option removes all highlighted endonucleases from the library of available endonucleases. The user is prompted to confirm deletion before it happens.
7.3 Sequences
The 'Sequences' menu includes options for managing sequence selection for use in analyses and sequence assignment to groups for use in group based analyses. Display of XML fileds associated with INSDSeq XML files can also be accessed here.
7.3.1 Select all. This selects all sequences currently loaded from a file for use in any analysis.
7.3.2 Select range. This selects a range sequences highlighted by the user for use in any analysis.
7.3.3 Deselect all. This deselects all sequences.
7.3.4 Deselect range. This deselects a range of sequences.
7.3.5 View and edit sequences. This opens a new window in which the sequences in memory are displayed graphically (an example is shown below). This window has its own set of menus providing options for editing sequences and their names.
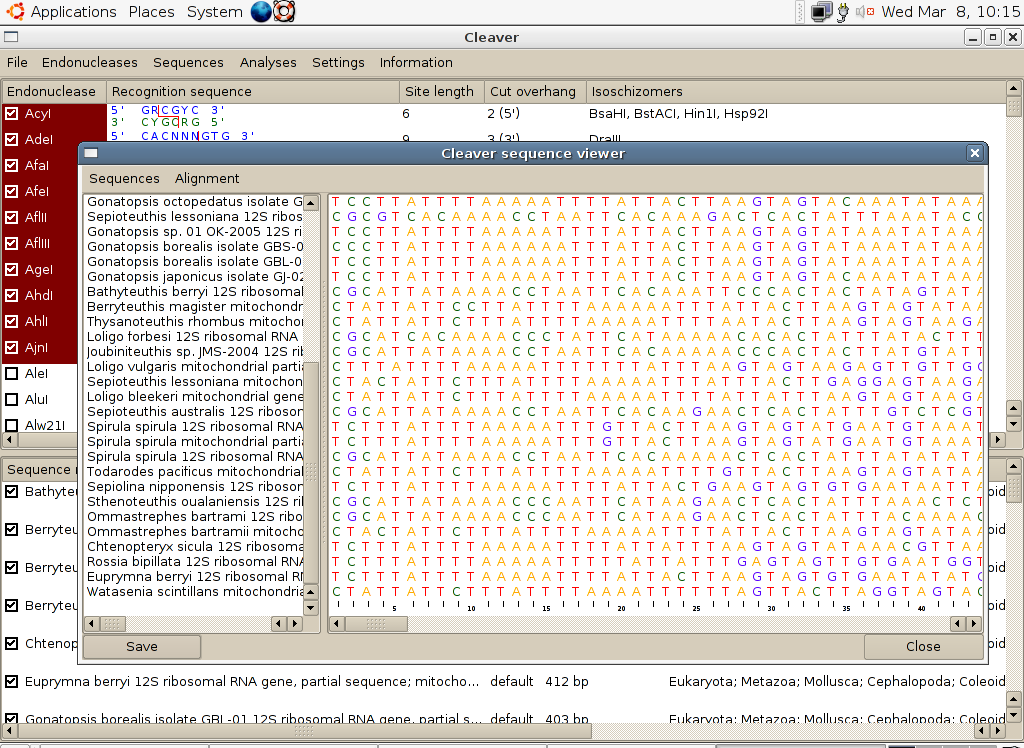
The menu options for the sequence editor are:
......7.3.5.1 Cut sequences. This cuts one or more sequences, selected by their names, from the alignment.
......7.3.5.2 Paste sequences. This pastes any previously selected sequences back into the alignment at a point selected from the list of names.
......7.3.5.3 Reverse. This reverses the sequences selected so that the nucleotides are shown in the reverse order (3' – 5' instead of 5' – 3').
......7.3.5.4 Complement. This converts sequences selected to the sequence complementary to the original sequence.
......7.3.5.5 Reverse complement. This converts the sequences selected to the antisense of the original, sense sequences.
......7.3.5.6 Search for oligonucleotide binding sites. This brings up a dialog that accepts entry of one or two oligonucleotides. The left hand entry box is for a 'forward' PCR primer that binds to the antisense strand. The right hand box is for a 'reverse' primer that binds to the sense strand. Cleaver searches the selected sequence for binding sites for these oligos, searching across gaps created by alignment of the sequences if this has happened. Degenerate primers can be entered and all possible binding sites for them will be recognised.
......7.3.5.7 Edit name for sequence. Opens a dialog for editing the name of the sequence.
......7.3.5.8 Edit sequence. Allows the sequence to be edited base-by-base.
......7.3.5.9 Remove columns. Allows a range of residues from all sequences in the alignment to be removed.
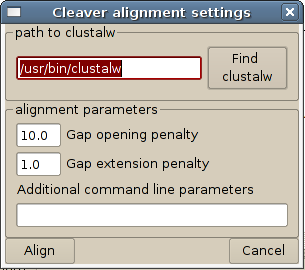
......7.3.5.10 Align sequences. This opens another dialog for aligning sequences using the program clustalw (shown below). To make the alignment process work, Cleaver must know where clustalw is located on the user's computer. The place where Cleaver will look for clustalw is indicated in the window that comes up when the 'Align sequences' option is selected. If Cleaver cannot find clustalw, an error message will be returned and alignment will not happen until the user directs Cleaver to the correct location. This location can be changed either here, or in the preferences options. Alignment gap opening and gap extension parameters can also be changed here.
7.3.6 Define groups manually. This allows the user to assign groups of sequences or individual sequences to a group with an arbitrary name. This is useful when the user wants to find endonucleases that cut one group of sequences, but not another.
7.3.7 Assign groups by taxonomy. Groups are assigned to sequences based on the taxonomic information included in INSDSeq XML files. The user must select the taxonomic level at which groups are to be assigned.
7.3.8 Show another INSDSeq XML field. If the sequences in memory were loaded from a INSDSeq XML file, this option allows display of information in that file that is not displayed by default.
7.4 Analyses
The analyses menu lists the analysis options.
7.4.1 Single digest of all selected sequences. All selected sequences are digested with all selected endonucelases in separate 'reactions' and the results are given per endonuclease as a list of sequences followed by the sizes of the fragments produced by digestion with that enzyme. The results can be saved as a text file using the button at the bottom of the window.
7.4.2 Multiple digests of each selected sequence. All selected sequences are digested with all selected endonucelases in one multi-enzyme reaction. The results are given as a list of sequences and a list of fragments that might be produced by a digest with the given enzyme combination. The results can be saved a s a text file.
7.4.3 Cut frequency analysis. The number of times that each selected endonuclease cutes each selected sequence is given. The results can be saved as a text file.
7.4.4 5' terminal restriction fragments. This analysis provides a list of the 5' most fragments produced by digesting the selected sequences with each selected endonuclease. The lists are given in descending order of fragment size. A summary of the proportions of fragments that do not have recognition sites and the proportion of fragments that are non-unique in size is given for each endonuclease. A summary of the informativeness of each endonuclease is given at the end of the results, where the proportion of non-unique fragments produced by each endonuclease for the selected sequences is given.
7.4.5 3' terminal restriction fragments. As above, but for the 3' terminal fragments.
7.4.6 Restriction maps – detailed sequence. This produces a set of overlapping restriction maps for all selected sequences with all selected endonucleses so that the maps can be compared between sequences. The restriction maps are in text format and show every base in the sequence. Note that if the default font for displaying results is changed to a non-monospaced font in the 'Preferences' dialog, this option will produce messy results. A monospaced font like 'courier' is necessary for the sequences to line up properly. The results can be saved as a text file.
7.4.7 Restriction maps – graphical overview. This shows each sequence in memory as a line with the relative positions of endonuclease recognition sites indicated on the map. Multiple seqeunces are shown on one page. The picture can be saved as a portable network graphic (.png) file that is readable by most graphics editing packages. This function is not available in the Windows version of Cleaver due to a problem with PyQt on Windows.
7.4.8 Search for group-specific endonucleases. If there are several groups of sequences defined, the user can search for endonucleases that cut one group, but not another using this option.
7.5 Settings
The settings menu has checkboxes for hiding for options that are used in all analyses. It also has options for manipulating Cleaver's preferences that are saved to file and loaded when the program starts up.
7.5.1 Linear DNA. When this is checked, DNA sequences are assumed to be linear. Unchecking it automatically checks the crcular DNA option. This is the default option.
7.5.2 Circular DNA. DNA sequences are assumed to be circular when this is checked.
7.5.3 Sort fragments by size. When selected, this default option means that DNA fragments resulting from endonuclease digests are sorted from largest to smallest. The alternative option is to return results in their natural order on undigested DNA.
7.5.4 Show fragments in 5' – 3' cut order. Fragments produced by restriction digestion are reported in the order that they occur on the undigested DNA, the 5' most fragment being reported first and the 3' most one last.
7.5.5 Preferences. Cleaver settings that persist between runs of the program can be edited here. A tabbed dialog containing these setting is given and those that are edited can be saved. This is especially useful for fine-tuning the fonts and their sizes used by your computer as these vary between the display managers used by different operating systems. Cleaver is designed to run on as many platforms as possible, but may need a little fine tuning to look good on your specific platform.
7.5.6 Restore default preferences. Restores a set of preferences that should allow Cleaver to work reasonably well. You are likely to want to edit the preferences to make the graphics Cleaver uses work as well as possible on your system.
7.6 Information
The information menu.
7.6.1 About cleaver. This re-displays the copyright statement that is displayed on starting Cleaver.
7.6.2 About Qt. This gives some information on the graphical user interface that Cleaver uses.
7.6.3 Online manual. This starts a web browsing programme and to this manual, which is stored in HTML format at sourceforge.net. This manual can also be saved so that it can be viewed without a web connection. The MacOSX version of Cleaver simply opens a web browser. There is some Mac-specific problem with passing command line arguments to a new process that the author cannot solve.
8. Troubleshooting
Cleaver is designed to run on all common personal computer systems. As a result, there are inevitable platform-specific peculiarities in its operation. The most likely problems to be encountered are getting Cleaver to interact with clustalw or a web browser. The locations that Cleaver thinks these programs are in are given in the preferences dialog and can be altered if they are wrong. If this is the case, Cleaver will tell you that something is wrong. There is a button labelled 'find clustalw' in the alignment dialog. Pressing this will allow you to search your harddrive for clustalw and when found, the preferences will be updated and saved for future sessions.
Another problem may be that fonts are too small or large on a specific computer, or are unavailable. The font choices can also be edited in the preferences dialog. The font for displaying results should always be a monotype font (with even spacing for all characters) while the font for displaying sequences in the sequence editor can be any type of font.
9. References
Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Research, 22:4673-4680.
![]()